 One of the best set of resources we have for bioinformatics, and especially microbiome research, are the extensive and freely available DNA sequence archives. For the past few years, most studies have been (and in most cases required to) archiving their relevant sequence datasets so that they are freely available to the public and other researchers. This is becoming an increasingly valuable resource for data mining and meta-analyses now that we have about a decade of archiving behind us. Just as these datasets can be highly valuable research tools, they can also be particularly difficult resources to download and prepare for analysis. I have been meaning to get to this for a while, so this week I want to go through an introduction to downloading these datasets. My goal is to equip you to easily get the sequence sets onto your own computer and start your own analysis.
One of the best set of resources we have for bioinformatics, and especially microbiome research, are the extensive and freely available DNA sequence archives. For the past few years, most studies have been (and in most cases required to) archiving their relevant sequence datasets so that they are freely available to the public and other researchers. This is becoming an increasingly valuable resource for data mining and meta-analyses now that we have about a decade of archiving behind us. Just as these datasets can be highly valuable research tools, they can also be particularly difficult resources to download and prepare for analysis. I have been meaning to get to this for a while, so this week I want to go through an introduction to downloading these datasets. My goal is to equip you to easily get the sequence sets onto your own computer and start your own analysis.The Sequence Read Archive (SRA)
One of the largest (if not the largest) sequence dataset archives available to the public is the United States National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA). This sequence archive has years of DNA sequencing studies readily available, but getting the reads can be a little bit of a challenge. They do have instructions (and other tools for downloading) in their documentation, but to make things easier, we will go through it here while including some custom scripts that you can use.
An easy way to get SRA datasets using command line tools is downloading the data from their ftp (no worries if you don't know what that is; it's just a site to download data from). As long as you are downloading a small-ish dataset, the wget tool works great. A nice subroutine you can use is as follows.
DownloadFromSRA () { line="${1}" echo Processing SRA Accession Number "${line}" mkdir ./data/${Output}/"${line}" shorterLine=${line:0:3} shortLine=${line:0:6} echo Looking for ${shorterLine} with ${shortLine} # Recursively download the contents of the wget -r --no-parent -A "*" ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByStudy/sra/${shorterLine}/${shortLine}/${line}/ mv ./ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByStudy/sra/${shorterLine}/${shortLine}/${line}/*/*.sra ./data/${Output}/"${line}" rm -r ./ftp-trace.ncbi.nih.gov }
export -f DownloadFromSRA
If you copy and paste this into your command line (Linux/Mac), you can just type the subroutine name "DownloadFromSRA", followed by the project ID that you want to use, and it will download all of the samples for you. If you are using a Mac, be sure to install wget using something like Homebrew (which I highly suggest for downloading tools in general). The files you get will be in the SRA format, so you have to remember to convert them to fastq format using their custom tools.
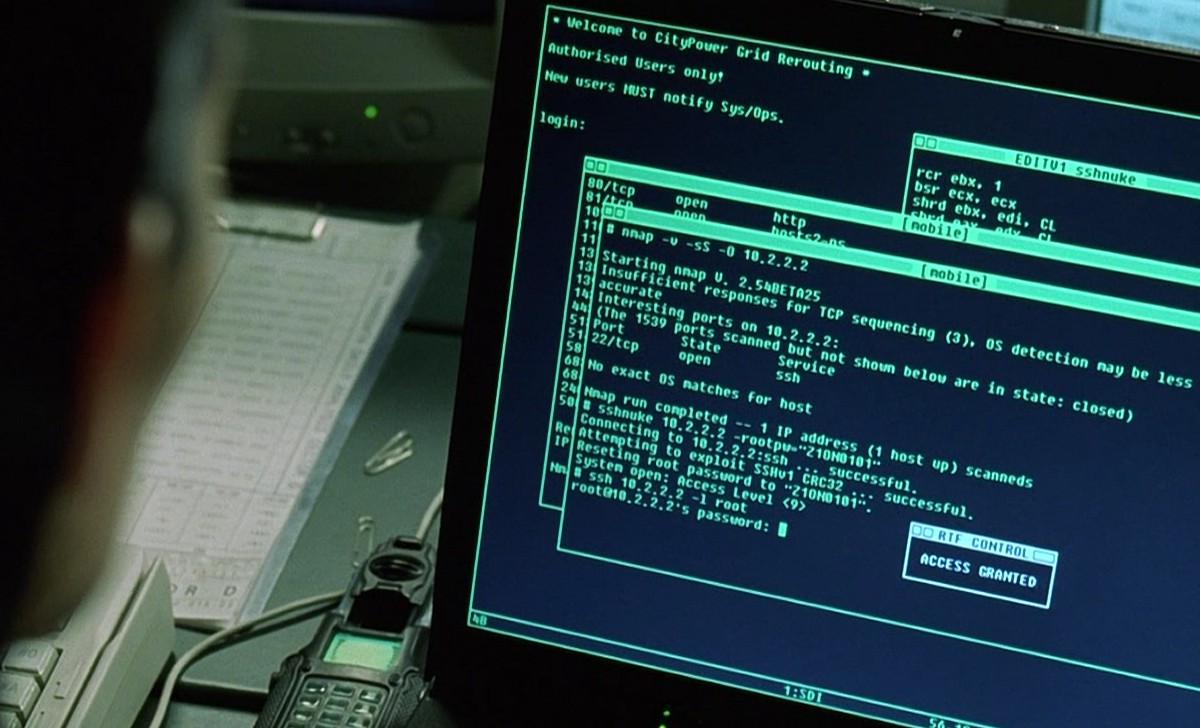
 |
| You don't have to be a superhero hacker to get DNA data from public archives. |
The Metagenomics RAST Server (MG-RAST)
Although used less than the SRA, the Metagenomics RAST Server (MG-RAST) is another one of the major archives available for free public use. Although MG-RAST is a nice sequence repository, it is unfortunately more difficult to use than the SRA (for downloading sequences at least). The key to downloading MG-RAST data with command line tools is honestly complicated at first, and sort of hidden in the documentation. Again, to make things easier, we can use some custom scripts to make things happen.
The trick to getting the MG-RAST sequence files using a project ID is that you have to first download the project metadata, and then use the parsed metadata information to download the actual files (this is done in the second loop below. The actual URL to use with their API is also kind of confusing, but once you get it you are ready to go.
DownloadFromMGRAST () { line="${1}" echo Processing MG-RAST Accession Number "${line}" mkdir -p ./data/"${line}" # Download the raw information for the metagenomic run from MG-RAST wget -O ./data/"${line}"/tmpout.txt "http://api.metagenomics.anl.gov/1/project/mgp${line}?verbosity=full" # Pasre the raw metagenome information for indv sample IDs sed 's/metagenome_id\"\:\"/\nmgm/g' ./data/"${line}"/tmpout.txt \ | sed 's/\".*//' \ | grep mgm \ > ./data/"${line}"/SampleIDs.tsv # Get rid of the raw metagenome information now that we are done with it rm ./data/"${line}"/tmpout.txt # Now loop through all of the accession numbers from the metagenome library while read acc; do echo Loading MG-RAST Sample ID is "${acc}" # file=050.1 means the raw input that the author meant to archive wget -O ./data/"${line}"/"${acc}".fa "http://api.metagenomics.anl.gov/1/download/${acc}?file=050.1" done < ./data/"${line}"/SampleIDs.tsv # Get rid of the sample list file rm ./data/"${line}"/SampleIDs.tsv } export -f DownloadFromMGRAST
These files will be in the fasta format instead of the sra format you get from the SRA. Also note that this uses GNU sed, which is not installed on Mac computers by default (Mac has a different version of sed. I know, it's kind of annoying). So make sure that, if you are running this on a Mac, install GNU sed using Homebrew again.
To give it a try, copy and paste this subroutine into your command line, and then write the project ID, like below.
DownloadFromMGRAST 4843
Conclusions
So there you have it. A very brief introduction to downloading SRA and MG-RAST datasets, with an emphasis on providing you the tools to do it yourself. Go ahead and give it a try. Let me know how it works, and if you run into problems, feel free to reach out with questions. And of course, please let me know if you have any questions, comments, or concerns!
Finally, thanks for reading! If you are a frequent reader, you might have noticed that my posts have been less frequent lately. I apologize for that. This has been an eventful year, which is great in general but bad for keeping up with the blog. As usual, it means I have some other exciting projects going on, and I am excited to share those experiences on here later. So for now the posts will be less frequent, but I look forward to getting back in a more frequent writing groove in the near future.
With the whole digital revolution, i usually argue that there should be a software engineer in every house. I myself am quite intrigued with programming and this was helpful.
ReplyDeleteProphage: A Primer On Ing Sequencing Data From Mg-Rast And The Sra >>>>> Download Now
Delete>>>>> Download Full
Prophage: A Primer On Ing Sequencing Data From Mg-Rast And The Sra >>>>> Download LINK
>>>>> Download Now
Prophage: A Primer On Ing Sequencing Data From Mg-Rast And The Sra >>>>> Download Full
>>>>> Download LINK 3H
This was helpful. Thanks!
ReplyDeletecall +2348038253815 or add us on whatsApp +2348038253815 or email illuminaticult0666@gmail.com GREETINGS!!!!! FROM THE GREAT GRAND MASTER! IN REGARDS OF YOU BECOMING A MEMBER OF THE GREAT ILLUMINATI, WE WELCOME YOU. Be part of something profitable and special (WELCOME TO THE WORLD OF THE ILLUMINATI). Are you a POLITICIAN, ENGINEER,DOCTOR, ENTERTAINER,MODEL,GRADUATE/ STUDENT,OR YOU HAVE IT IN MIND TO EXPAND YOUR BUSINESS/COMPANIES TO BECOME GREAT MINDS. It is pertinent to also know that For becoming a member, you earn the sum of $1,000,000 as the illuminati membership salary monthly.Be a part of this GOLDEN “OPPORTUNITY” The great illuminati Organization makes you rich and famous in the world, it will puxll you out from the grass root and take you to a greater height were you have long aspired to be and together we shall rule the world with the great and mighty power of the Illuminati, long life and prosperity here on earth with eternal life and jubilation. You can reach Us on illuminaticult0666@gmail.com
ReplyDeleteHello everyone..Welcome to my free masterclass strategy where i teach experience and inexperience traders the secret behind a successful trade.And how to be profitable in trading I will also teach you how to make a profit of $7,000 USD weekly and how to get back all your lost funds feel free to Email: (carlose78910@gmail.com )
ReplyDeleteVia whatsapp: (+12166263236)
God is Good!
ReplyDeleteI promised God that I would share my testimony on this blog. I had all the signs of STD Virus but I was not too sure as to which one. I did a lot of online research and scared myself straight for a whole week before going to see the nurse. She took one look at my genital part and first said that it could just be the anatomy of my body, then she said it looked like genital warts and that I may have herpes. I was devastated. She gave me some medicine for the herpes and some cream for the warts. I was also tested for every single STD including herpes. I went home and cried searching the web for all sorts of cures for herpes and awaiting my results. I saw a post whereby Dr. Oyagu cured Herpes and other diseases, I copied his contacts out and added him on whats app via (+2348101755322). The next day my test result was ready and i confirmed Herpes positive. I told Dr.Oyagu about my health problems and he assured me of cure. He prepared his herbal medicine and sent it to me. I took it for 14 days (2 weeks). Before the completion of the 14 days in which I completed the dose, the Blisters and Warts that were on my body was cleared. I went back for check-up and I was told I'm free from the virus. Dr. Oyagu cures all types of diseases and viruses with the help of his herbal medicine. You can reach Dr. Oyagu via his email address on (oyahuherbalhome@gmail.com) or WhatsApp him on (+2348101755322) Visit His website on https://oyaguspellcaster.wixsite.com/oyaguherbalhome
Prophage: A Primer On Ing Sequencing Data From Mg-Rast And The Sra >>>>> Download Now
ReplyDelete>>>>> Download Full
Prophage: A Primer On Ing Sequencing Data From Mg-Rast And The Sra >>>>> Download LINK
>>>>> Download Now
Prophage: A Primer On Ing Sequencing Data From Mg-Rast And The Sra >>>>> Download Full
>>>>> Download LINK
Denizli
ReplyDeleteKonya
Denizli
ısparta
Bayburt
GJFL
whatsapp görüntülü show
ReplyDeleteücretli.show
71MA
görüntülü.show
ReplyDeletewhatsapp ücretli show
34BSS
https://titandijital.com.tr/
ReplyDeletebalıkesir parça eşya taşıma
eskişehir parça eşya taşıma
ardahan parça eşya taşıma
muş parça eşya taşıma
İAF41
kocaeli evden eve nakliyat
ReplyDeletekilis evden eve nakliyat
bursa evden eve nakliyat
trabzon evden eve nakliyat
hakkari evden eve nakliyat
XTMUES
C4D88
ReplyDeleteMuş Lojistik
Bursa Evden Eve Nakliyat
Gümüşhane Lojistik
Aksaray Parça Eşya Taşıma
Bursa Lojistik
A283D
ReplyDeleteBitfinex Güvenilir mi
Çankırı Şehir İçi Nakliyat
Bingöl Lojistik
Bitcoin Nasıl Alınır
Ankara Şehir İçi Nakliyat
Kocaeli Şehir İçi Nakliyat
İzmir Evden Eve Nakliyat
Muğla Evden Eve Nakliyat
Ünye Parke Ustası
97B15
ReplyDeletebuy winstrol stanozolol
Ankara Asansör Tamiri
https://steroidsbuy.net/steroids/
testosterone enanthate
Burdur Evden Eve Nakliyat
buy deca durabolin
buy dianabol methandienone
Çankırı Evden Eve Nakliyat
buy primobolan
43B3D
ReplyDeleteÇorum Parça Eşya Taşıma
Ünye Marangoz
Tekirdağ Boya Ustası
Kars Lojistik
Çerkezköy Marangoz
Karabük Lojistik
Silivri Fayans Ustası
Çerkezköy Çekici
Çorlu Lojistik
7ACDE
ReplyDeleteTrabzon Şehirler Arası Nakliyat
Etlik Fayans Ustası
Cointiger Güvenilir mi
Kırklareli Evden Eve Nakliyat
Trabzon Şehir İçi Nakliyat
Sonm Coin Hangi Borsada
Mercatox Güvenilir mi
Niğde Şehirler Arası Nakliyat
Osmo Coin Hangi Borsada
5750E
ReplyDeleteBilecik Sohbet Siteleri
Sivas Yabancı Canlı Sohbet
en iyi rastgele görüntülü sohbet
Tokat Telefonda Canlı Sohbet
ankara nanytoo sohbet
mersin rastgele görüntülü sohbet
burdur görüntülü sohbet sitesi
adana chat sohbet
antalya chat sohbet
115E0
ReplyDeleteAdana Parasız Görüntülü Sohbet Uygulamaları
Aydın Yabancı Görüntülü Sohbet Uygulamaları
Ağrı Canli Sohbet
Kırşehir Canlı Ücretsiz Sohbet
Adana Canlı Sohbet Odaları
karaman rastgele sohbet odaları
bilecik canlı sohbet siteleri
Kırklareli Görüntülü Sohbet Siteleri
düzce ücretsiz sohbet siteleri
BDAD6
ReplyDeleteİpsala
Çamoluk
Şemdinli
Başiskele
Delice
Karasu
Derbent Bayanlar
Kuluncak
Buharkent