After a couple awesome years and over 100,000 views, Prophage is moving to a new site. Check out the first post on the new site here. Follow the link below for the new site.
New Prophage Blog Site
Saturday, November 18, 2017
Saturday, June 17, 2017
Improving Your Skill Set: Tips for Learning New Programming Languages
 I spend a considerable amount of my time with scientists who are staring to learn to code (either IRL or online), often in the hopes that it will open future research and career doors. One of the major barriers I have found in my own experience, as well as observed with others, is learning and implementing new programming languages after I already learned one. If we are honest, learning a programming language is very challenging but also incredibly rewarding. So do we really want to go through that again with a new language, and will the new language open as many analytical doors as the first? This week I was to discuss the process of learning new programming languages, and offer you some tips on learning a new language, if that is something that interests you.
I spend a considerable amount of my time with scientists who are staring to learn to code (either IRL or online), often in the hopes that it will open future research and career doors. One of the major barriers I have found in my own experience, as well as observed with others, is learning and implementing new programming languages after I already learned one. If we are honest, learning a programming language is very challenging but also incredibly rewarding. So do we really want to go through that again with a new language, and will the new language open as many analytical doors as the first? This week I was to discuss the process of learning new programming languages, and offer you some tips on learning a new language, if that is something that interests you.Monday, May 8, 2017
A Primer on Downloading Sequencing Data from MG-RAST & the SRA
 One of the best set of resources we have for bioinformatics, and especially microbiome research, are the extensive and freely available DNA sequence archives. For the past few years, most studies have been (and in most cases required to) archiving their relevant sequence datasets so that they are freely available to the public and other researchers. This is becoming an increasingly valuable resource for data mining and meta-analyses now that we have about a decade of archiving behind us. Just as these datasets can be highly valuable research tools, they can also be particularly difficult resources to download and prepare for analysis. I have been meaning to get to this for a while, so this week I want to go through an introduction to downloading these datasets. My goal is to equip you to easily get the sequence sets onto your own computer and start your own analysis.
One of the best set of resources we have for bioinformatics, and especially microbiome research, are the extensive and freely available DNA sequence archives. For the past few years, most studies have been (and in most cases required to) archiving their relevant sequence datasets so that they are freely available to the public and other researchers. This is becoming an increasingly valuable resource for data mining and meta-analyses now that we have about a decade of archiving behind us. Just as these datasets can be highly valuable research tools, they can also be particularly difficult resources to download and prepare for analysis. I have been meaning to get to this for a while, so this week I want to go through an introduction to downloading these datasets. My goal is to equip you to easily get the sequence sets onto your own computer and start your own analysis.Saturday, April 8, 2017
Publication Alert: High Nucleotide Resolution Study of the Skin Virome
 |
| We identified diversity generating retroelements as a potential mechanism driving targeted genomic diversity. |
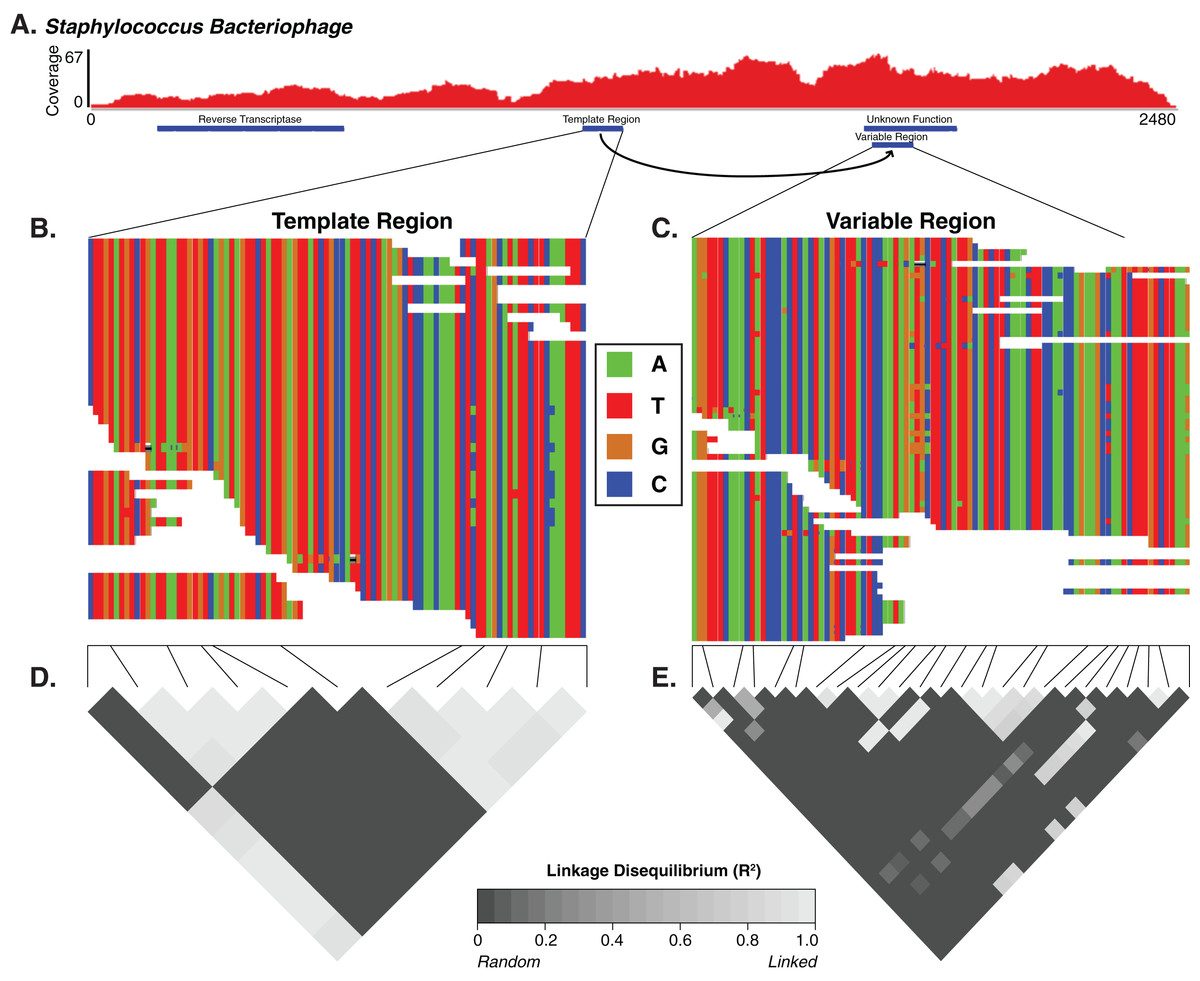
A few weeks ago some colleagues and myself published a new manuscript looking at the diversity of the human skin virome. In our previous previous work, we evaluated the diversity of viruses on the skin. Other groups have looked at virus diversity at other body sites including the gut, lungs, and oral cavity. Our new paper focused on the diversity within viruses on the skin. It provided initial insight into the genomic variability associated with major viruses in the skin virome. In other words, it was a "high resolution" study of the virome.
Sunday, March 12, 2017
Correlations In Random Genomic Data: A Simple Biology Pitfall
 Wow it has been a long time since we have had a post on here! As always, that means other projects are in the works and the blog has taken a bit of a back seat, but we are back and ready to talk science. This week I wanted to get to a topic I have been meaning to get to for a while. If you are a frequent reader, you know that every now and then I like to go over some basic statistics topics that cause confusion among biologists, as well as scientists in other fields. This week I want to cover a common statistical pitfall, with the hopes that it will prevent readers from making simple mistakes. The topic for this post will be obtaining statistically significant correlations from random gene expression data.
Wow it has been a long time since we have had a post on here! As always, that means other projects are in the works and the blog has taken a bit of a back seat, but we are back and ready to talk science. This week I wanted to get to a topic I have been meaning to get to for a while. If you are a frequent reader, you know that every now and then I like to go over some basic statistics topics that cause confusion among biologists, as well as scientists in other fields. This week I want to cover a common statistical pitfall, with the hopes that it will prevent readers from making simple mistakes. The topic for this post will be obtaining statistically significant correlations from random gene expression data.Saturday, January 28, 2017
A Model for Phage Communication and the Implications for the Human Microbiome
 |
| The research group prepared two types of media to test phage infection efficacy. |
Well we took a bit of a break these past couple of weeks, but we are back for the new year! Welcome to the Prophage blog 2017! The year has actually been off to a good start, with a lot of interesting papers being published this January. This week I want to kick things off by covering a very cool 2017 study by Erez et al that described an new and interesting mechanism by which bacteriophages communicate using their bacterial hosts. This really is a well written and elegant study that I highly suggest you read. In this post, I want us to cover the highlights of the study, and then discuss what this will mean for future research endeavors.
The research group led by Erez et al began their work by testing the hypothesis that "bacteria secrete communication molecules to alert other bacteria of phage infection", but what they ended up finding was arguably much more interesting. They began their series of experiments by simply growing bacteria in liquid media with and without bacteriophages (see the figure to the right). They let the mixture sit long enough for the phages to infect their bacterial hosts for a couple of replication cycles (3 hours), and then removed all of the bacteria and phages from the liquid by filtration. At this point, if there was a signaling molecule released during the infection, it would still be in the media even though the phages and bacteria were removed. Additionally, if there was a signaling molecule released during the phage infection period, repeating an infection in that same media would result in altered growth patterns (for example, less bacteria killed when the molecule is present). As it turns out, this is exactly what they observed.
Sunday, December 11, 2016
How to Write a Manuscript Submission Cover Letter
 The communication of our research findings is a foundational pillar to our careers as scientists. One of the most common ways we scientists share information is by publishing papers in peer-reviewed journals. This primary method of information dissemination allows us to share our research findings both to our colleagues as well as the public at large. When preparing a manuscript for submission to a journal for peer review and subsequent publication, a lot of work goes into preparing a variety of documents. One of the important documents is a cover letter to the editor. This letter represents a significant hurdle for new and young researchers because it is often unclear what a cover letter should actually look like, and what information should be included. In this week's post I want to go over what a good cover letter could look like and how you can write your own. I say this is what it could look like because there is certainly a lot of room for interpretation and personal style, and there are many correct ways to do it. Here I am just going to cover one potential way to tackle the problem.
The communication of our research findings is a foundational pillar to our careers as scientists. One of the most common ways we scientists share information is by publishing papers in peer-reviewed journals. This primary method of information dissemination allows us to share our research findings both to our colleagues as well as the public at large. When preparing a manuscript for submission to a journal for peer review and subsequent publication, a lot of work goes into preparing a variety of documents. One of the important documents is a cover letter to the editor. This letter represents a significant hurdle for new and young researchers because it is often unclear what a cover letter should actually look like, and what information should be included. In this week's post I want to go over what a good cover letter could look like and how you can write your own. I say this is what it could look like because there is certainly a lot of room for interpretation and personal style, and there are many correct ways to do it. Here I am just going to cover one potential way to tackle the problem.
Subscribe to:
Posts (Atom)