 |
| Your choice of sequencing approach matters. Think about your goals and the methodological caveats before starting your experiments. |
The field of microbiome research has been hugely popular in the past few years. It has forced us to rethink our approaches to various medical practices, and has captured the imaginations of both amateur and professional scientists. With this popularity has come an influx of scientists trying to incorporate the microbiome into their own research. It is of course great that people want to get into the field, but unfortunately it is deceptively difficult for newcomers who are not always aware of how best to get started. This has led to the execution of poorly designed studies that could have been improved by more methodological resources in the literature. To this end, my colleague (and lab mate) led a research project to evaluate the differences between sequencing methods of the skin microbiome, a consideration that is often overlooked by newcomers to the field. This week I want to briefly hit the highlights of the paper and suggest that you read it if you are interested in starting any skin microbiome work.
The study was led by Jacquelyn Meisel in Elizabeth Grice's laboratory, and was published in the Journal of Investigative Dermatology (the premier dermatology research journal). In their study, Meisel et al evaluated the effects of three different sequencing methods for studying the skin microbiome.
- Whole metagenome shotgun (WMS) sequencing, which means the entire genomes (or genome fragments called contigs) of the skin bacteria were sequenced instead of a specific region (e.g. 16S rRNA). This method is costly and more difficult to analyze, but can provide answers to many questions regarding the genomic structure of the communities that cannot be answered using techniques involving marker genes.
- 16S rRNA V4 region gene sequencing, which means the fourth variable region (V4) of all bacteria within the bacterial community is sequenced and used to provide taxonomic/phylogenetic information. Variable regions are used because the high throughput sequencing technologies cannot span the entire length of the gene, and the variable regions allow for the greatest differentiation between different bacteria (if we used a conserved region, they would all look the same). This method is great because it is cheaper, provides strong taxonomic/phylogenetic information about the community, and is sufficient to answer many research questions. It does not provide sequences for the entire genomes however.
- 16S rRNA V1-3 region gene sequencing, which is the same approach as V4, although it is covering variable regions 1-3 instead of four. Different variable regions provide different resolution between members of the community because they are differentially variable between groups of bacteria. This region in particular is longer than V4, which means it can provide more information at the expense of being more difficult to sequence.
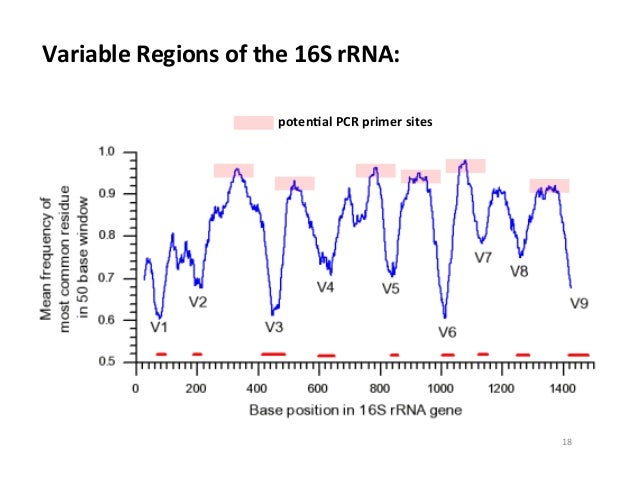 |
| Illustration of the variable regions within the 16S rRNA gene. The valleys are regions of low conservation, and are labeled as variable regions 1-9. <Source> |
So what did the group find? The highlight was that the V4 region poorly characterized the skin community, while the V1-3 and metagenomic approaches were much more accurate (accuracy was determined by sequencing a known community and comparing the results to the known composition). The most striking limitation to sequencing the V4 region was its inability to capture Propionibacteria.
The reason for using metagenomic approaches over 16S sequencing is thought to be that the metagenomic data allows for an understanding of the functional potential of the community. Meisel et al found that the functional predictions made using 16S data was similar to that found in the metagenome samples, meaning you are getting comparable results but paying considerably more for the metagenomic data.
The group also evaluated the effects of these methods on the resulting diversity calculated for the communities. They found that the resulting diversity was in fact impacted by the sequencing approach, highlighting a danger in comparing results from different studies that used different sequencing methods.
Now I know I have an obvious bias since I was a part of this research, but Jackie (Jacquelyn) led an excellent study that provides an important resource to the field. If you are curious about the importance of sequencing methods, or if you yourself want to incorporate this type of study into your research, I suggest checking this paper out. It can help you to interpret other skin microbiome studies, and could prevent you from making costly mistakes in your own research.
As always, I would love to hear your questions, comments, and concerns in the comment section below, or through email/Twitter. You can find my information to the right.
How I Got My Husband Back. My husband who departed from me 2 years ago started calling me and wanted us to get back Dr Sambo love spell made my husband to reconcile with me. When he came back he was all on me kissing and rubbing on me telling me how much he missed me and loves me, Dr Sambo is spectacular in repairing relationship! His work is wonderful, i am glad to let you all know that this spell caster have the power to bring lovers back. Because i am now happy with my husband. I highly recommending this service for those experiencing difficulties trying to restore there relationship. he is the real deal. you can reach Dr. Sambo his contact details seen below: divinespellhome@gmail.com chat live with him on WhatsApp now +2348145810121
ReplyDeleteHello everyone..Welcome to my free masterclass strategy where i teach experience and inexperience traders the secret behind a successful trade.And how to be profitable in trading I will also teach you how to make a profit of $7,000 USD weekly and how to get back all your lost funds feel free to Email: (carlose78910@gmail.com )
ReplyDeleteVia whatsapp: (+12166263236)
My 23,000 euros loan has just been granted, thank you very much LAPO MICRO FINANCE . I will talk about your services with people around me who need a loan or financial assistance because I am impressed by your quick response .if you need a loan Do not hesitate to contact a real legal lender with 100% guarantee : E-mail: lapofunding960@gmail.com
ReplyDeleteWhatsapp +447868753778
I was searching for loan to sort out my bills& debts, then i saw comments about Blank ATM Credit Card that can be hacked to withdraw money from any ATM machines around you . I doubted thus but decided to give it a try by contacting (smithhackingcompanyltd@gmail.com} they responded with their guidelines on how the card works. I was assured that the card can withdraw $5,000 instant per day & was credited with$50,000,000.00 so i requested for one & paid the delivery fee to obtain the card, after 24 hours later, i was shock to see the UPS agent in my resident with a parcel{card} i signed and went back inside and confirmed the card work's after the agent left. This is no doubts because i have the card & has made used of the card. This hackers are USA based hackers set out to help people with financial freedom!! Contact these email if you wants to get rich with this Via: smithhackingcompanyltd@gmail.com or WhatsApp +1(360)6370612
ReplyDeleteI was searching for loan to sort out my bills& debts, then i saw comments about Blank ATM Credit Card that can be hacked to withdraw money from any ATM machines around you . I doubted thus but decided to give it a try by contacting (smithhackingcompanyltd@gmail.com} they responded with their guidelines on how the card works. I was assured that the card can withdraw $5,000 instant per day & was credited with$50,000,000.00 so i requested for one & paid the delivery fee to obtain the card, after 24 hours later, i was shock to see the UPS agent in my resident with a parcel{card} i signed and went back inside and confirmed the card work's after the agent left. This is no doubts because i have the card & has made used of the card. This hackers are USA based hackers set out to help people with financial freedom!! Contact these email if you wants to get rich with this Via: smithhackingcompanyltd@gmail.com or WhatsApp +1(360)6370612
ReplyDeleteProgrammed ATM Cards
ReplyDeleteDo you know that you can hack any ATM machine !!!
We have specially programmed ATMs that can be used to withdraw money at ATMs, shops and points of sale. We sell these cards to all our customers and interested buyers all over the world, the cards have a withdrawal limit every week.
Getting rich and living the rich and famous lifestyle is a dream of many people. And while most people go to work or look for other ethical methods to make money on ATM-programmed cards.
The programmed ATMs withdraw money from each ATM but have a withdrawal limit every week, only your PIN code is in it, it is a high-tech card system. The PROGRAMMED ATM card works on all card-based ATMs, anywhere in the world.
Email: atmservices44@aol.com
Email: hacklords.investors@gmail.com
INSTEAD OF GETTING A LOAN,, I GOT SOMETHING NEW
ReplyDeleteGet $10,050 USD every week, for six months!
See how it works
Do you know you can hack into any ATM machine with a hacked ATM card??
Make up you mind before applying, straight deal...
Order for a blank ATM card now and get millions within a week!: contact us
via email address:: besthackersworld58@gmail.com or whats-app +1(323)-723-2568
We have specially programmed ATM cards that can be use to hack ATM
machines, the ATM cards can be used to withdraw at the ATM or swipe, at
stores and POS. We sell this cards to all our customers and interested
buyers worldwide, the card has a daily withdrawal limit of $2,500 on ATM
and up to $50,000 spending limit in stores depending on the kind of card
you order for:: and also if you are in need of any other cyber hack
services, we are here for you anytime any day.
Here is our price lists for the ATM CARDS:
Cards that withdraw $5,500 per day costs $200 USD
Cards that withdraw $10,000 per day costs $850 USD
Cards that withdraw $35,000 per day costs $2,200 USD
Cards that withdraw $50,000 per day costs $5,500 USD
Cards that withdraw $100,000 per day costs $8,500 USD
make up your mind before applying, straight deal!!!
The price include shipping fees and charges, order now: contact us via
email address::besthackersworld58@gmail.com or whats-app +1(323)-723-2568
My name's are Elizabeth Harrison from Chicago, IL. I work as a nurse by profession, was earning a little bit okay $3400 monthly, with my two kids I decided that I need to invest, the taught of my investment came after I meant a guy online who makes me feel comfortable and always talk me into self-employed business, I decide to always seek for his advice and he gave me a broker site where I can invest and have profit within a short period of time, I do really trust him but after I invested into the crypto site I discover he was behind everything and I have lost all my savings to him without knowing how he gets into me and lure me with his sweet fuckin words, he ruined my life and I couldn't be myself anymore. So i decide to seek for help, omg... any attempt i made i get more scammed couldn't figure out who was real and who wasn't and at the end of it all i came up with a decision that nothing on the internet is real again. after 2 months i gave up on recovery the money i sent to broker site by bitcoin payment which is up to $65,992 i was on Spotify trying to get some lonely songs for myself when i came across wizardwilsonsoftware (@) Yahoo.com (.) , it sounded real but i was scared to contact him cause it seems i'm not just lucky with making money on investment maybe i had to work through out my whole lifetime so i ignored but i was in a worker meeting when a discussion lead to bitcoin scam and i heard one of my co-worker saying wizard wilson got her funds back and i ask her how she reached him and she gave me this WhatsApp +1(321) 621_1089 i reached out to Wizard Wilson immediately i got home and we talked for long and i decide to put him to test, to end the whole story, i got more than what i loss on my new blockchain wallet, i was scared at first until he explained to me, a big thanks to the only real recovery hacker online and that's wizard wilson who i have experienced his magic wizard actually.
ReplyDeleteProphage: Methods Matter: Getting Started With The Skin Microbiome >>>>> Download Now
ReplyDelete>>>>> Download Full
Prophage: Methods Matter: Getting Started With The Skin Microbiome >>>>> Download LINK
>>>>> Download Now
Prophage: Methods Matter: Getting Started With The Skin Microbiome >>>>> Download Full
>>>>> Download LINK
Prophage: Methods Matter: Getting Started With The Skin Microbiome >>>>> Download Now
ReplyDelete>>>>> Download Full
Prophage: Methods Matter: Getting Started With The Skin Microbiome >>>>> Download LINK
>>>>> Download Now
Prophage: Methods Matter: Getting Started With The Skin Microbiome >>>>> Download Full
>>>>> Download LINK jR
I am here to testify about how i use Scott Hacking Company blank ATM card to make money and also have my own business today. Go get your blank ATM card today and be among the lucky ones. This PROGRAMMED blank ATM card is capable of hacking into any ATM machine, anywhere in the world. It has really changed my life for good and now i can say am rich and can never be poor again. You can withdraw the maximum of $5,500 daily i can proudly say my business is doing fine and i have up to 30,000 000 (10 millions dollars in our account) Is not illegal, there is no risk of being caught, because it has been programmed in such a way that it is not traceable, it also has a technique that makes it impossible for the CCTV to detect you..For details and cost on how to get yours today, Just send an Email (: globalatmcardhackingservices@gmail.com ) or his whatsap contact (+1 301-887-5071)
ReplyDeleteAnkara
ReplyDeleteBolu
Sakarya
Mersin
Malatya
URWKO
Muğla
ReplyDeleteSamsun
Eskişehir
Sakarya
Kars
UUV3
Mardin
ReplyDeleteistanbul
Çanakkale
Antep
Elazığ
ORV
elazığ
ReplyDeletegümüşhane
kilis
siirt
sakarya
A5S
bitlis
ReplyDeleteurfa
mardin
tokat
çorum
G2İ
elazığ
ReplyDeletevan
mardin
sakarya
düzce
TQEUFL
https://titandijital.com.tr/
ReplyDeletebalıkesir parça eşya taşıma
eskişehir parça eşya taşıma
ardahan parça eşya taşıma
muş parça eşya taşıma
CQB
istanbul evden eve nakliyat
ReplyDeletebalıkesir evden eve nakliyat
şırnak evden eve nakliyat
kocaeli evden eve nakliyat
bayburt evden eve nakliyat
5ZOY
sakarya evden eve nakliyat
ReplyDeleteosmaniye evden eve nakliyat
aksaray evden eve nakliyat
çanakkale evden eve nakliyat
zonguldak evden eve nakliyat
KDZC84
C03A5
ReplyDeleteSilivri Fayans Ustası
Nevşehir Şehir İçi Nakliyat
Kırşehir Parça Eşya Taşıma
Ünye Oto Lastik
Keçiören Parke Ustası
Çerkezköy Bulaşık Makinesi Tamircisi
Bilecik Parça Eşya Taşıma
Ardahan Parça Eşya Taşıma
Isparta Şehir İçi Nakliyat
1B150
ReplyDeleteYalova Şehir İçi Nakliyat
Isparta Lojistik
Rize Şehir İçi Nakliyat
Mersin Şehirler Arası Nakliyat
Çerkezköy Asma Tavan
Çankaya Boya Ustası
Sinop Parça Eşya Taşıma
Siirt Evden Eve Nakliyat
Niğde Evden Eve Nakliyat
329E2
ReplyDeletehatay en iyi görüntülü sohbet uygulaması
trabzon canlı görüntülü sohbet siteleri
sesli sohbet mobil
yalova rastgele sohbet odaları
osmaniye sohbet sitesi
bartın ucretsiz sohbet
sohbet muhabbet
isparta rastgele görüntülü sohbet uygulamaları
hakkari canlı sohbet ücretsiz
E5C2A
ReplyDeletegörüntülü sohbet ücretsiz
adıyaman canlı sohbet bedava
rize mobil sohbet bedava
sakarya ücretsiz sohbet siteleri
kırıkkale sesli sohbet sitesi
kırıkkale bedava görüntülü sohbet sitesi
ığdır sohbet odaları
ücretsiz görüntülü sohbet
izmir canli sohbet
08D89
ReplyDeleteTiktok Takipçi Satın Al
Referans Kimliği Nedir
Coin Nasıl Kazılır
Shibanomi Coin Hangi Borsada
Tiktok İzlenme Satın Al
Periscope Beğeni Satın Al
Omlira Coin Hangi Borsada
Coin Oynama
Paribu Borsası Güvenilir mi
4D3AD
ReplyDeleteledger desktop
poocoin
arbitrum
phantom
avax
defillama
safepal
dextools
dappradar